
Corona: Den genetischen Ursprüngen auf der Spur
Einer Gruppe internationaler Forschender aus der Genetik und Archäologie aus Kiel und Cambridge ist es durch Anwendung phylogenetischer Netzwerkanalysen gelungen, den Ursprung und die Verbreitung des neuartigen Coronavirus (SARS-CoV‑2) nachzuvollziehen, das die Lungenkrankheit COVID-19 verursacht. Es ist die erste große, von Expertinnen und Experten begutachtete und damit qualitätsgesicherte Analyse der Entwicklung der vollständigen Erbinformationen des Virus in diesem weltweiten Ausbruch.

Beteiligt waren Wissenschaftlerinnen und Wissenschaftler um Dr. Michael Forster, Institut für Klinische Molekularbiologie (IKMB) des Universitätsklinikums Schleswig-Holstein (UKSH), Campus Kiel, und der Christian-Albrechts-Universität zu Kiel (CAU), ebenso wie das Team von Dr. Peter Forster vom McDonald Institute for Archaeological Research an der Universität Cambridge.
Ihre am Mittwoch, 8. April 2020, in der renommierten Fachzeitschrift PNAS veröffentlichten Ergebnisse zeigen, dass sich in Europa und Amerika andere Virus-Typen verbreiten als in China.
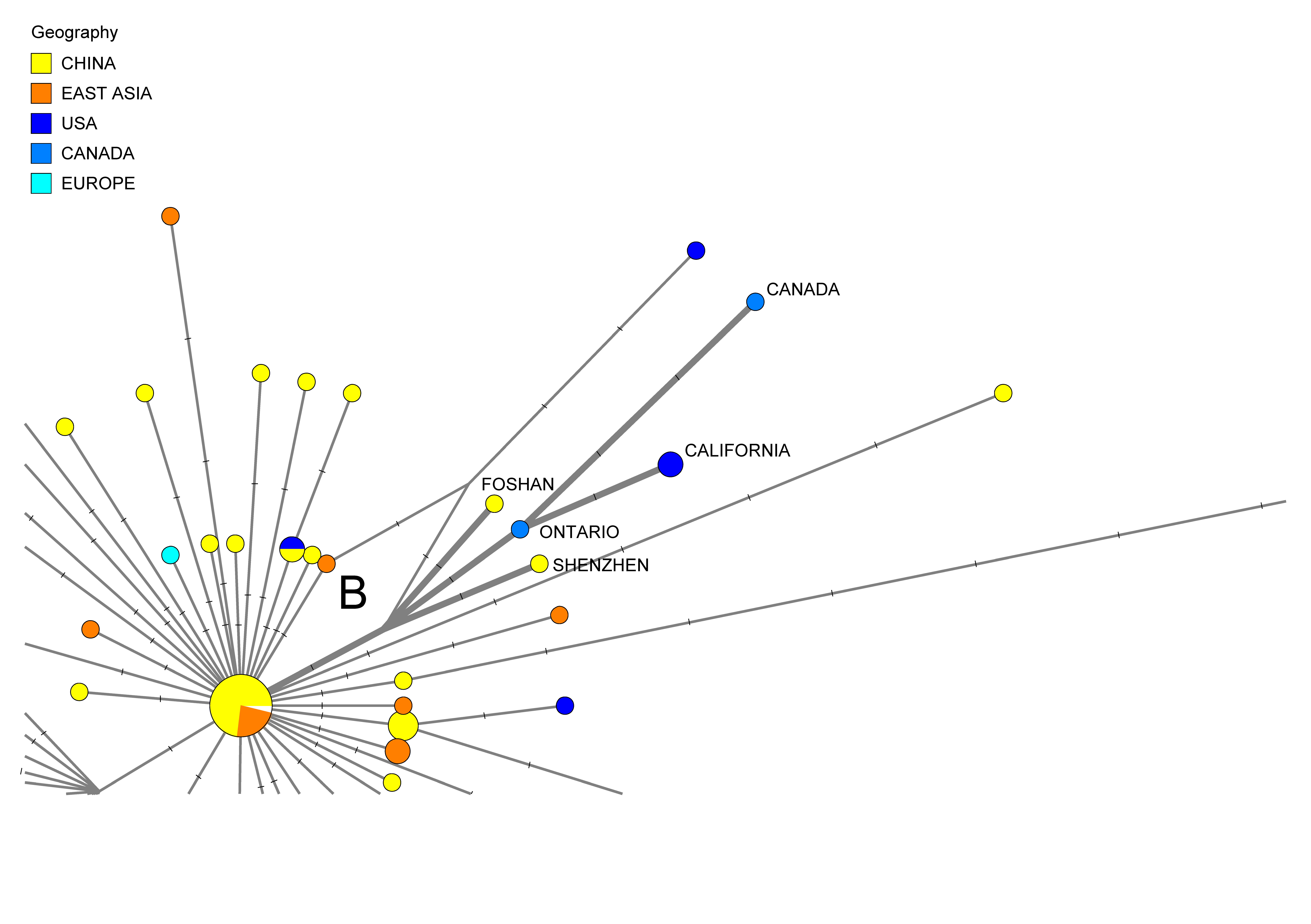
Bevor SARS-CoV‑2 im letzten Jahr einen Menschen infizierte, hatten sich bereits Vorfahren dieses Virus in tierischen Wirten entwickelt. Nach heutigem Erkenntnisstand ist ein Fledermaus-Coronavirus dem menschlichen SARS-CoV‑2 am ähnlichsten. Dies geht aus dem Vergleich der Virus-Erbinformation, dem sogenannten Virusgenom, hervor. Die Forschenden verglichen weiterhin in einer sogenannten phylogenetischen Netzwerkanalyse, d.h. in einer Untersuchung der genetischen Verwandtschaftsverhältnisse, die ersten 160 vollständigen Genome der menschlichen COVID-19-Viren, die zu Beginn des aktuellen Ausbruchs von Ende 2019 bis März 2020 von den weltweiten Forschungslaboren gesammelt wurden. Hierbei fanden die Forschenden drei zentrale Varianten, die sie als A, B und C bezeichnet haben. Typ A ist dem eng verwandten Fledermaus-Coronavirus am ähnlichsten und somit wahrscheinlich der Urahn aller menschlichen Coronaviren. Dies haben weitere Vergleiche mit zwei entfernter verwandten Pangolin-Coronavirus-Stämmen bestätigt. Interessanterweise ist der in Wuhan vorherrschende Typ B nicht der ursprüngliche menschliche Virustyp. Aber auch in Wuhan kommt Typ A, also das ursprüngliche menschliche Virusgenom, durchaus vor.
Über Phylogenetische Netzwerkmethoden:
Phylogenetische Netzwerkmethoden dienen der Untersuchung der Entwicklung und Verbreitung einer Spezies (oder Art) eines Lebewesens oder Virus (die nicht zu Lebewesen gezählt werden). Die ersten dieser Methoden wurden 1979 in Neuseeland etabliert. In den 1990er Jahren wurden sie von deutschen Forschenden der Mathematik unabhängig weiterentwickelt, vor allem von Prof. Hans-Jürgen Bandelt und Dr. Arne Röhl in Hamburg in Zusammenarbeit mit dem Kieler Wissenschaftler Dr. Peter Forster, damals am Heinrich-Pette-Institut für Virologie in Hamburg. 1998 entdeckte der renommierte Archäologie-Professor Colin Renfrew (Cambridge) die Vorteile der Analysemethode und gründete 1999 mit Dr. Peter Forster die weltweit erste archäogenetische Forschungsgruppe in einem archäologischen Institut. Forster war Gründungsmitglied der Jungen Akademie und wurde 2012 zum Mitglied der Nationalen Akademie der Wissenschaften (Leopoldina) berufen. Er kooperiert eng mit dem Kieler Institut für Klinische Molekularbiologie. Im Kieler Medical Life Sciences Studiengang gehört die Netzwerkmethode daher zum Erstsemester-Lehrinhalt von Prof. Dr. Almut Nebel.
In dieser ersten Phase des Ausbruchs waren die A- und C‑Typen in signifikanten Anteilen außerhalb Ostasiens zu finden – bei Betroffenen in Europa, Australien und Amerika. Im Gegensatz dazu ist der B‑Typ der häufigste Typ in Ostasien. Der C‑Typ ist unter anderem früh in Singapur dokumentiert worden und ist auch unter den ersten europäischen Infektionsfällen häufig vertreten.
Die Forschenden verwendeten eine sogenannte phylogenetische Netzwerkanalyse. Diese Methode war ursprünglich in der archäologischen Forschung zur Rückverfolgung der Vorgeschichte der menschlichen DNA und zur Rekonstruktion prähistorischer Sprachen entwickelt worden. Im Gegensatz zu konventionellen Stammbaummethoden ermöglicht es dieser Ansatz, hunderte mögliche Stammbäume gleichzeitig in einer übersichtlichen Darstellung anzuzeigen. Dies ist ein wesentlicher Vorteil, wenn der wahre Stammbaum unbekannt ist.
Die Anwendung dieser Methode im Fall des Coronavirus bedeutet, dass die Infektionswege für dokumentierte COVID-19-Fälle genau nachgezeichnet werden können: So wurde beispielsweise zunächst angenommen, dass der erste norditalienische Infektionsfall („Patient Eins“) von einer bestimmten Wuhan-Kontaktperson aus seinem Bekanntenkreis infiziert worden war. Doch als diese Kontaktperson getestet wurde, stellte sich heraus, dass sie das Virus nicht hatte. Die Suche nach dem italienischen „Patienten Null“ endete somit in einer Sackgasse und eine wirksame Quarantäne potenziell infizierter Personen war unmöglich. Seitdem hat sich die Krankheit unkontrolliert in Italien ausgebreitet. Das phylogenetische Netzwerk weist auf mindestens zwei unabhängige frühe Infektionswege in Italien hin, von denen einer mit dem ersten bekannten Fall in Deutschland und der andere mit dem Ausbruch im sogenannten „Singapur-Zweig“ in Verbindung steht. Die phylogenetische Rückverfolgung könnte daher künftig dabei helfen, COVID-19-Infektionsquellen ungeklärter Herkunft zu identifizieren, die dann zur Eindämmung künftiger Ausbrüche der Krankheit unter Quarantäne gestellt werden können.
„Unsere Ergebnisse unterstreichen die Notwendigkeit der genomischen Sequenzierung und der Anwendung der Methode der phylogenetischen Netzwerkanalyse“, sagt IKMB-Wissenschaftler Dr. Michael Forster, der die Studie in Zusammenarbeit mit dem von der Deutschen Forschungsgemeinschaft (DFG) geförderten deutschlandweiten Gensequenzierungs-Zentrum „Competence Centre for Genomic Analysis“ (CCGA) Kiel an der CAU durchführte. „Wir sind überrascht, dass für Italien, einem der am frühesten und stärksten betroffenen EU-Länder, trotz der hervorragenden Forschenden bisher nur eine Handvoll italienischer Fälle in der globalen COVID-19-Falldatenbank GISAID gemeldet wurden“, fügt Prof. Andre Franke hinzu.
Originalpublikation: Peter Forster, Lucy Forster, Colin Renfrew, Michael Forster (2020):
Phylogenetic network analysis of SARS-CoV‑2 genomes. Proceedings of the National Academy of Sciences First published 08.04.2020 DOI: https://doi.org/10.1073/pnas.2004999117
Textquelle: Oliver Grieve, Universitätsklinikum Schleswig-Holstein
Bildquellen: Verbreitungszweige der verschiedenen SARS-CoV-2-Varianten, Foto: Michael Forster, Uni Kiel / Dr. Michael Forster, Institut für Klinische Molekularbiologie (IKMB) des UKSH, Campus Kiel, Foto: Christian Urban, Uni Kiel